Our paper titled “Utilizing molecular simulation, ideal adsorbed solution theory and ensemble learning algorithms to investigate adsorption and separation of sulfides on amorphous nanoporous materials” has been published online in the journal “Next Materials”, accessible at https://doi.org/10.1016/j.nxmate.2024.100378
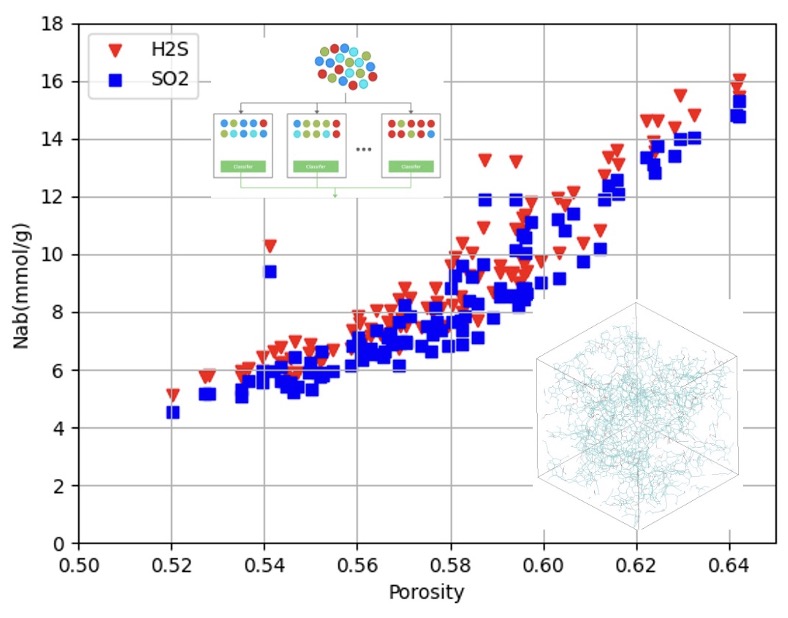
Using grand canonical Monte Carlo simulations, we studied the adsorption of pure H2S and SO2 gases on amorphous materials and the separation of CH4-H2S and CO2-SO2 mixtures. At 303 K, HCP-Colina-id016 was the best adsorbent for both gases, with 16 mmol/g. For CH4-H2S, aCarbon-Marks-id002 had the highest selectivity (~80) but low H2S adsorption (~1 mmol/g), while Kerogen-Coasne-id013 showed higher H2S adsorption (12 mmol/g) with a selectivity of 20. For CO2-SO2, HCP-Colina-id018 had a SO2 selectivity over 30 and SO2 adsorption of 12 mmol/g. IAST underestimated adsorption and selectivity, but molecular simulations showed CO2 condensation caused a drop in SO2 adsorption. Ensemble learning models ranked as XGBoost > GBDT > RF > CatBoost for predicting H2S and SO2 adsorption.
Next Materials is a peer-reviewed, open access journal under Elsevier’s Next family, focused on materials science. It offers speed, consistency, and easy submission for authors, while providing readers with seamless access. The journal aims to be a key resource for both academic and industry researchers, showcasing innovative and influential work in materials science. Managed by a dedicated in-house editorial team, Next Materials serves as a platform for inspiring the next generation of materials scientists.